-
Kill Dogma Revolution !

 .
.L’infezione alla luce della biologia strutturale
https://sibbm.zanichelli.it/italiano/2020/...ia-strutturale/
Introduzione
La pandemia di COVID-19 attualmente in corso ha urgentemente spinto l’intera comunità scientifica a dedicare ernomi sforzi, lavoro e risorse all’identificazione e allo sviluppo di nuove strategie farmacologiche per arrestarel’infezione da SARS-CoV-2 (di seguito indicato con CoV-2). Come nell’arte della guerra, per poter sconfiggere il nemico è fondamentale conoscere: com’è fatto il virus, qual è la sua forma? Come infetta le cellule umane? Come cresce, replica e si sviluppa nelle cellule ospite? Di che cosa ha bisogno per sopravvivere? Rispondere a tali domande fornisce le armi, le informazioni chiave a cui i ricercatori ambiscono per sviluppare vaccini e farmaci antivirali sicuri ed efficaci.
A molte di queste domande è già stato risposto. Ma come si possono ottenere queste informazioni senza poter vedere il nemico? Una particella virale e tutto il macchinario molecolare che usa per replicarsi e sopravvivere nelle cellule ospite non è né visibile a occhio nudo né usando un classico microscopio ottico. È qui che entra in gioco la biologia strutturale, il cui scopo è proprio quello di identificare la struttura tridimensionale delle macromolecole biologiche, come le proteine e gli acidi nucleici, e di correlarla con la loro funzione fisiopatologica. Questa disciplina scientifica si basa su tecniche estremamente avanzate che consentono di visualizzare e analizzare molecole invisibili e di combattere invisibili agenti patogeni. La conoscenza della forma, della struttura tridimensionale, e quindi della chimica del CoV-2 e di tutte le biomolecole responsabili dell’infezione e della replicazione virale costituisce il mezzo ottimale per sviluppare strategie di cura e per identificare, progettare e produrre molecole come vaccini e farmaci antivirali, che bersaglino e arrestino le sue funzioni e i suoi effetti dannosi sulla salute umana.
Al momento, le strutture di 13 delle 26 proteine di CoV-2 sono state depositate nella banca dati internazionale delle strutture di proteine (Protein Data Bank, PDB), ma ci si aspetta che il numero cresca rapidamente nel prossimo futuro.
Nei prossimi paragrafi, partendo dalla descrizione dell’architettura complessiva del virus, verranno descritte le strutture delle tre protein di CoV-2 che sono considerate attualmente come i migliori bersagli farmacologici potenziali, ovvero la proteasi principale Mpro, la proteina Spike (S) e l’RNA polimerasi dipendente dall’RNA (RNA dependent RNA polymerase, RdRp). Verrà anche analizzata la relazione che lega le strutture di queste molecule alle loro funzioni fisiopatologiche. Le tecniche di biologia strutturale citate sono descritte nella sezione Approfondimenti: le tecniche.
CoV-2: un altro coronavirus con la corona
Il primo isolamento documentato del CoV-2 a partire da campioni prelevati di pazienti infetti è stato realizzato all’ospedale Spallanzani di Roma1 e ha permesso di intraprendere lo studio del nuovo agente patogeno virale in diversi laboratori a livello internazionale. Le immagini delle particelle virali, isolate da persone di tutto il mondo, sono state ottenute usando un microscopio particolate che sfrutta elettroni anziché fotoni come sorgente di radiazione, ossia il microscopio elettronico a trasmissione (TEM). Le proprietà ottiche degli elettroni rendono possibile osservare oggetti fino a 1–10 000 volte più piccoli di 1 µm (10−6 m). Le immagini ottenute mediante TEM di CoV-2 hanno confermato che il virus appartiene alla famiglia dei coronavirus, data la tipica forma a corona della superficie esterna (Figura 1).
Ora sappiamo che il CoV-2 fa parte, più nello specifico, dei β-coronavirus, costituiti da un RNA a singolo filamento con senso positivo (v. Il mondo sorpredente del genoma di SARS-CoV-2), di circa 29,9 kilobasi (kb; 1 kilobase = 1000 basi). Una particella virale (il virione) di CoV-2 ha un nucleocapside composto dall’RNA genomico e ricoperto da proteine fosforilate che interagiscono con la membrana virale durante l’assemblaggio del virione, giocando un ruolo critico nel potenziare la replicazione del virus2. L’RNA genomico e il nucleocapside sono avvolti da un doppio strato di fosfolipidi in cui sono immerse diverse proteine che svolgono ruoli cruciali per l’infezione e la replicazione: la proteina S, la proteina di membrana (M), l’emoagglutinina esterasi (HE) e la proteina del rivestimento (E). La particella virale ha un diametro di 60–100 nm (10−9 m) e appare rotonda o ovale3.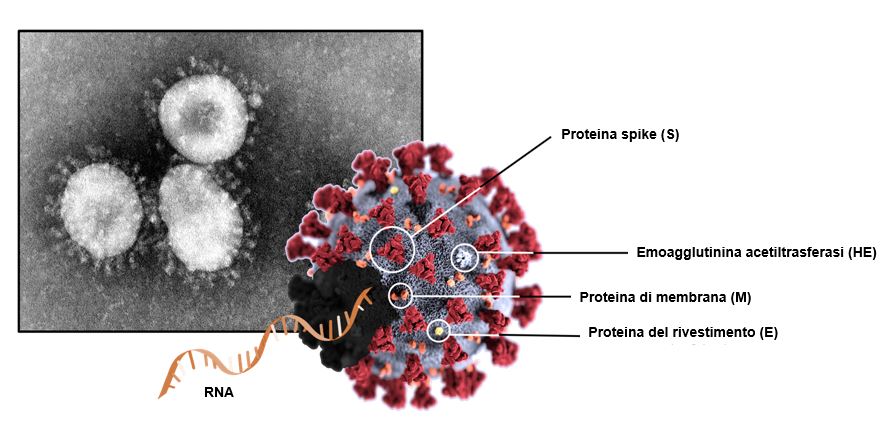
La proteasi principale del CoV-2: l’origine dell’intero arsenale molecolare del virus
Durante l’infezione della cellula ospite, il genoma virale agisce come RNA messaggero (v. Il mondo sorpredente del genoma di SARS-CoV-2) e dirige la sintesi di due grandi poliproteine (pp1a e pp1ab) che contengono al loro interno proteine più piccole necessarie alla produzione di nuove particelle virali all’interno delle cellule infette. Tale insieme di proteine comprende: un complesso di replicazione/trascrizione, diverse proteine strutturali necessarie a costruire virioni e due proteasi4,5. Queste due proteasi giocano un ruolo essenziale poiché tagliano le due grandi poliproteine nelle proteine funzionali più piccole.
La protease principale di CoV-2, che effettua il maggior numero di tagli, pesa 33,8 kDa e si chiama Mpro, altrimenti conosciuta come proteasi 3C-simile (simile alla chimotripsina). La Mpro è fondamentale per la replicazione virale ed è assente nelle cellule umane. Per questa ragione essa rappresenta un buon bersaglio per lo sviluppo di nuovi farmaci antivirali: il blocco delle sue funzioni sarebbe infatti letale per il virus, ma sicuro per gli esseri umani.
Il meccanismo di azione di Mpro è simile a quello di altre proteasi. Tutte le proteasi possiedono due residui amminoacidici chiave: un residuo attivatorio (di solito un’istidina, His) che rimuove protoni da un gruppo ossidrilico o tiolico della catena laterale di un secondo residuo (di solito una serina, Ser, o una cisteina, Cys) che agisce come potente nucleofilo, ossia un potente donatore di elettroni. Nell’Mpro, che è una proteasi in cisteina, la diade catalitica è costituita da His41 e Cys1456 (i numeri identificano la posizione all’interno della sequenza amminoacidica della proteina).
Il meccanismo di reazione con cui Mpro catalizza l’idrolisi della poliproteina virale è descritto in Figura 2.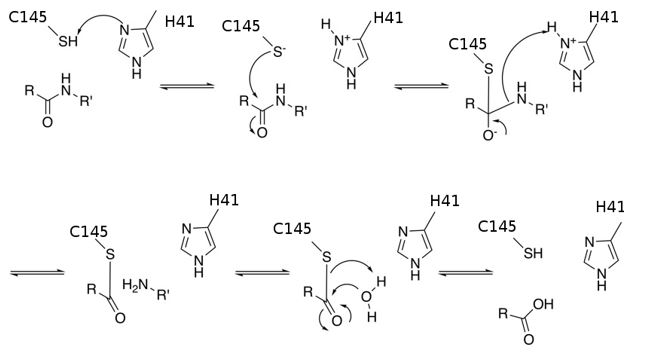
La struttura cristallografica della proteina Mpro in complesso con l’inibitore N3
Jin e collaboratori hanno ottenuto la struttura cristallografica ad alta risoluzione della proteina Mpro in complesso con N3 (Figura 3), una molecola nota per legare e inibire la proteasi principale di altri coronavirus, come SARS-CoV e MERS-CoV (codice di accesso in PDB: 6lu7)6. L’N3 è un inibitore simile a un oligopeptide (Figura 3D).
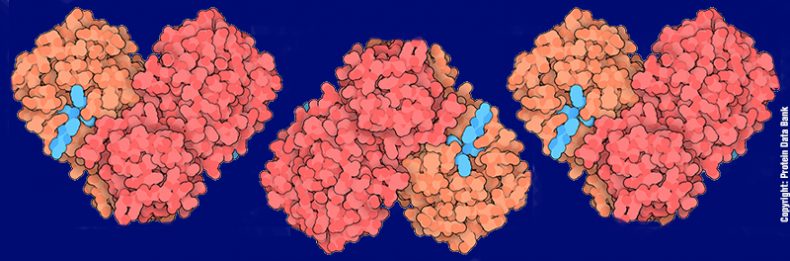
L’analisi della struttura mostra che la proteina appare come dimero (Figura 3A), formato da due subunità identiche di 306 aminoacidi (A e B). Ogni monomero è formato da tre differenti domini (Figura 3B):
dominio I (residui 1–101);
dominio II (residui 102–184);
dominio III (residui 201–306) .
I domini I e II hanno una tipica struttura a barile β in cui i filamenti β si dispongono in modo antiparallelo, mentreil dominio III è formato da cinque α-eliche. Il dominio III è unito al dominio II da un lungo ripiegamento (residui 185–200). Il sito di legame al substrato consiste in una cavità profonda che è posta in prossimità dell’interfaccia del dimero tra i domini I e II e contiene la diade catalitica Cys145-His41 (Figura 3C). L’analisi della struttura ha consentito di capire che l’inibitore N3 lega fortemente la cavità dell’Mpro che normalmente alloggia il substrato, formando un legame covalente con la Cys145. L’analisi dell’interazione tra N3 e i residui che rivestono la cavità hanno fornito importanti informazioni strutturali per progettare inibitori potenti e reversibili dell’Mpro.
Come la Mpro di SARS-CoV, quella di CoV-2 taglia le poliproteine pp1a e pp1ab in specifiche posizioni amminoacidiche, identificando i siti di taglio grazie a particolari “sequenze di base” nelle poliproteine7. Le posizioni dei residui che appartengono alle sequenze di base nelle poliproteine sono nominate a seconda della posizione relativa rispetto al sito di taglio ed è possibile identificarle in modo molto specifico (per i dettagli, vedere la legenda della Figura 3C e la referenza bibliografica n. 8).
La sovrapposizione di sequenze delle Mpro di 12 coronavirus, inclusi CoV-2, SARS-CoV e MERS-CoV, mostra che i residui che rivestono la tasca che lega il substrato sono fortemente conservati9-12. Questo suggerisce che un potente inibitore di CoV-2 potrebbe essere un composto leader per guidare la sintesi di farmaci ad ampio spettro contro tutte le infezioni da coronavirus.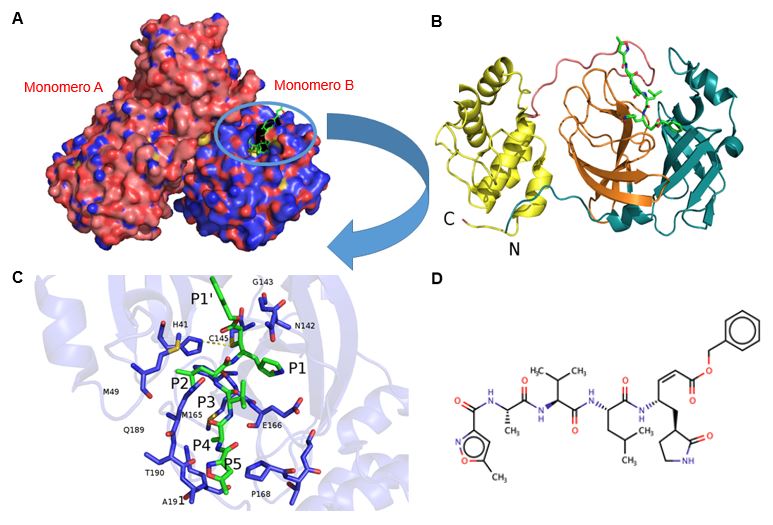
Dalla struttura dell’Mpro del CoV-2 ai farmaci: il ruolo degli screening virtuali e high-throughput
La struttura dell’Mpro di CoV-2 in complesso con l’inibitore N3 fornisce un modello e delle informazioni che possono essere usati per identificare altre molecole organiche capaci di legare la tasca catalitica dell’Mpro con più alta affinità, e condurre così allo sviluppo di nuovi farmaci antivirali specifici per il CoV-2. Una delle metodiche che è usata correntemente per identificare nuovi composti capostipiti è lo screening virtuale. Lo screening virtuale è una tecnica computazionale che permette di analizzare grandi numeri di dati (highthrouput screening) di librerie di migliaia di composti chimici, per identificare molecole che abbiano maggiori probabilità di legare il bersaglio farmacologico. Il legame del composto chimico al bersaglio viene simulato in silico (ossia in maniera totalmente predittiva), riducendo fortemente i costi, il tempo e lo sforzo rispetto allo stesso screening eseguito sperimentalmente. Una volta identificato un numero più piccolo di composti, vengono eseguiti esperimenti di laboratorio per confermare e ulteriormente selezionare il composto che lega a maggior affinità il bersaglio.
Jin e collaboratori6 hanno usato proprio questa tecnica per identificare molecole con un volume corrispondente a quello della tasca catalitica dell’Mpro, partendo dalla sua struttura cristallografica in complesso con N3. Tutti i composti, tipicamente piccole molecole organiche, conservate in una specifica banca dati, sono stati testati in silico per individuare quelli che, date le proprietà geometriche e chimiche, potessero essere potenzialmente in grado di legare la Mpro nella tasca catalitica. Lo screening dei composti viene effettuato tramite programmi dedicati. In questo caso è stato usato Glide (versione 8.2). Questa analisi ha portato all’identificazione di un composto, detto cinaserina, che ha mostrato il punteggio più alto e la modalità più sensata di legame alla tasca catalitica dell’Mpro. La cinaserina è un antagonista della serotonina molto ben caratterizzato, che è stato testato preliminarmente sugli esseri umani negli anni ’60 del Novecento e che ha mostrato di inibire l’Mpro di SARS-CoV. Di rilievo è il fatto che la cinaserina non è tossica per le cellule umane, mostrando una citotossicità del 50% a concentrazioni più alte di 200 μM, mentre è capace di inibire l’Mpro di CoV-2 a concentrazioni più basse (IC50=125 μM). Perciò, la cinaserina rappresenta un buon candidato come farmaco antivirale, che potrebbe essere ottimizzato per ottenere il farmaco nella sua forma finale13.
Infatti, una volta che un nuovo composto è identificato, viene inizialmente somministrato ad animali da esperimento, e successivamente agli esseri umani nel corso di sperimentazioni cliniche per testarne sicurezza ed efficacia. Possono volerci dai 10 ai 15 anni, o anche di più, per completare tutte le fasi di una sperimentazione clinica prima di giungere allo stadio di licenza di un nuovo farmaco. Una strategia alternativa per accelerare i tempi è quella di testare l’attività di molecole esistenti e già approvate per altre patologie. Jin e collaboratori hanno applicato questa strategia per trovare velocemente farmaci contro COVID-1914. Infatti, hanno analizzato oltre 10 000 composti come ligandi possibili della Mpro, inclusi farmaci già approvati, alcuni candidati per le sperimentazioni cliniche e anche dei prodotti naturali. Tra questi, hanno identificato un derivato del 5-fluorouracile, il Carmofur (1-hexylcarbamoyl-5-fluorouracil), che è risultato capace di legare ed inibire l’Mpro (Figura 4, a sinistra). Il Carmofour è stato già approvato come farmaco antitumorale ed è usato per trattare il cancro del colon retto fin dal 1980. Sempre, Jin e collaboratori hanno risolto la struttura cristallografica dell’Mpro in complesso con il Carmofour, scoprendo le basi molecolari dell’efficacia del composto. In particolare, l’acido grasso di Carmofour lega la Cys145 attraverso un legame covalente, inibendo l’enzima (Figura 4, a destra). Questo studio è particolarmente importante poiché dimostra che il Carmofour, un farmaco già approvato e commercialmente disponibile, potrebbe essere potenzialmente usato contro la COVID-19 e fornisce le basi strutturali per progettare nuovi e più potenti inibitori ad ampio spettro che potrebbero essere usati per trattare tutte le infezioni da coronavirus.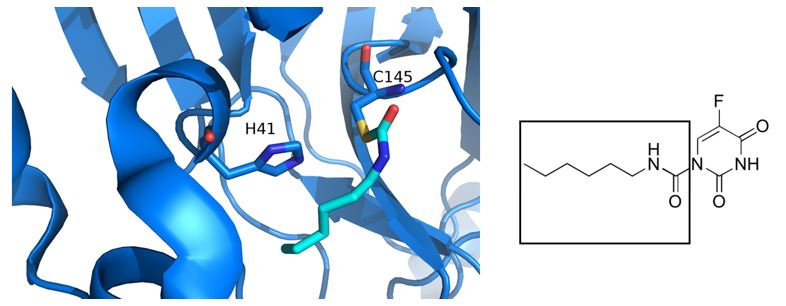
La proteina Spike del CoV-2: la chiave molecolare del virus
I virus evolvono continuamente le proteine della loro superficie per potenziare l’interazione con i recettori sulle cellule ed entrare in esse con maggior efficienza secondo il modello chiave-serratura. Questo è anche il caso della proteina Spike (S) di CoV-2 (la chiave) e del recettore umano Angiotensin Converting Enzyme 2 (hACE2, la serratura).
La proteina S è una delle più interessanti e studiate tra quelle che contribuiscono al legame con il recettore dell’ospite e alla patogenesi virale. La proteina S “decora” la superficie del virus ed è responsabile per l’aspetto a corona della superficie virale, da cui il nome coronavirus. Questa è usata dal virus come una chiave per entrare nelle cellule ospite15. Agisce legando il recettore sulle cellule bersaglio, induce l’endocitosi dei virioni e catalizza la fusione tra le membrane cellulari e virali, assicurando l’ingresso dell’RNA genomico virale nel citoplasma delle cellule. La proteina S rappresenta anche il bersaglio principale del sistema immunitario, attivandolo e inducendo la produzione di anticorpi. Per questa ragione è considerata il bersaglio primario di farmaci antivirali e vaccini. Fortunatamente, la mole di dati strutturali sulle proteine S dei coronavirus correlati al SARS-CoV (SARSr-CoVs, SARS-related CoVs), inclusa la struttura recentemente determinata di S di CoV-2 (d’ora in avanti indicata con CoV-2-S), costituiscono una ricca fonte di informazioni utili per progettare molecole inibitrici della sua funzione e, quindi, potenzialmente usabili intrattamenti terapeutici.
L’organizzazione strutturale di CoV-2-S è molto simile a quella delle proteina S di altri coronavirus come SARS-CoV e MERS-CoV. Essa è una proteina trimerica transmembrana formata da tre unità identiche, dette protomeri (Figura 5). Ogni protomero espone all’esterno delle catene di zuccheri che servono a ingannare il sistema immunitario16,17.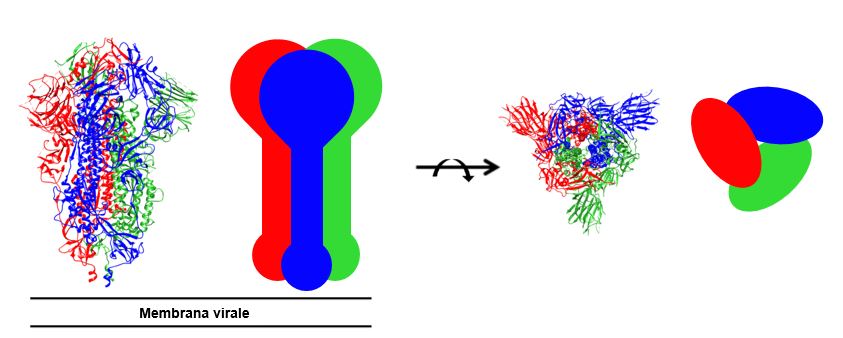
Ogni protomero di CoV-2-S (per esempio, quello blu nella Figura 5) comprende due subunità funzionali: una responsabile per il legame al recettore sulle cellule bersaglio (la subunità S1) e l’altra coinvolta nella fusione con la membrane cellulare (subunità S2). Più in dettaglio, la subunità S1 contiene:
un dominio N-terminale (NTD, N-Terminal Domain);
due subdomini detti SD1 e SD2 (Subdomain 1 e 2);
il dominio di legame al recettore (RBD, Receptor Binding Domain) responsabile per il legame alla cellula ospite attraverso l’interazione con ACE2 (vedi oltre);
un dominio C-terminale che contiene il macchinario di fusione che aiuta il virus ad entrare nelle cellule (Figura 6A)16,17.
La CoV-2-S, come varie proteine spike di altri SARSr-CoV, è tagliata da proteasi cellulari al confine tra le subunità S1 ed S2, generando due regioni separate che rimangono legate in modo non covalente nella cosiddetta “conformazione di prefusione”. Infatti, CoV-2-S esiste in due differenti conformazioni, chiamate “su” (up) e “giù” (down) (Figura 6B). Nella conformazione “giù”, la CoV-2-S non può mediare la fusione della CoV-2 con la membrana della cellula ospite. Per assicurare l’ingresso del virus, deve andare incontro ad un cambiamento conformazionale, che garantisce l’acquisizione della conformazione “su”16.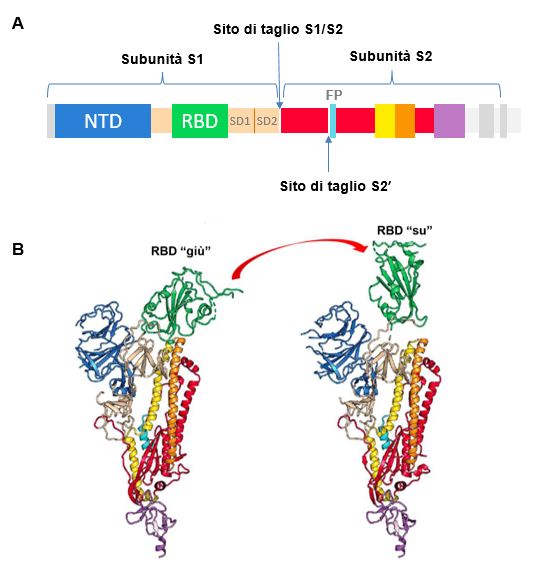
L’acquisizione della conformazione “su” si verifica non appena la CoV-2-S si avvicina ad ACE2; in seguito, avviene il taglio da parte delle proteasi cellulari nel sito S1/S2 (Figura 7). Dopo il taglio, le subunità S1 e S2 assolvono a differenti funzioni:
la S1 lega ACE2 attraverso l’RBD;
la S2 media l’ingresso del virus dopo un ulteriore taglio a un sito secondario chiamato S2′. Questo taglio si pensa attivi la fusione di membrane attraverso un altro esteso ed irreversibile cambiamento conformazionale.
In conclusione, l’ingresso del coronavirus in cellule suscettibili è un processo complesso che richiede l’azione concertata del legame al recettore e del processamento proteolitico della proteina S per promuovere la fusione tra virus e cellula. Una delle proteasi cellulari coinvolta nell’ingresso della CoV-2 nelle cellule ospiti è la serina proteasi transmembrana 2 (TMPRSS2, Transmembrane Serine Protease 2), che è richiesta anche per l’infezione da SARS-CoV. Infatti, l’inibizione di TMPRS22 blocca l’ingresso di SARS-CoV e CoV-2 nelle cellule.
È interessante il fatto che la CoV-2-S possieda un’inserzione di quattro amminoacidi al confine tra S1 ed S2 rispetto a alla proteina S di SARS-CoV. Questi quattro amminoacidi addizionali costituiscono il sito di taglio per una specifica proteasi umana chiamata furina16. La presenza di questo sito di taglio peculiare per la furina in CoV-2-S ha fatto ipotizzare che, data l’espressione praticamente ubiquitaria delle proteasi simili alla furina, esse potrebbero aver partecipato all’acquisizione del più ampio tropismo cellulare e tissutale di CoV-2 rispetto a SARS-CoV, come anche a un incremento della sua trasmissibilità e patogenicità16. È stato dimostrato di recente che, come TMPRSS2, la furina sia essenziale per l’ingresso di CoV-2 nelle cellule ospiti. Inoltre anche la catepsina D, una proteasi tipica dei lisosomi, è richiesta per un ingresso efficiente del CoV-2. Inoltre, è di rilievo che tutte queste proteasi sembrino cooperare per mediare l’infezione da CoV-2. Questo non è il caso di SARS-CoV, che non ha un sito di taglio per la furina.
Continua nel sito sopra , link...
 .
.

 Kill Dogma Tv-Nella Tana del Bianconiglio-Illuminati&Nuovo Ordine Mondiale NWO-Il manuale del risveglio-Ricerca-Indagini-Notizie
Kill Dogma Tv-Nella Tana del Bianconiglio-Illuminati&Nuovo Ordine Mondiale NWO-Il manuale del risveglio-Ricerca-Indagini-Notizie  BANCHE DATI NO ALLA SPERIMENTAZIONE CLANDESTINA-Fonti&Prove:Vaccini,Microchip,Transumanesimo,5G,Mondo Smart City Intelligenti
BANCHE DATI NO ALLA SPERIMENTAZIONE CLANDESTINA-Fonti&Prove:Vaccini,Microchip,Transumanesimo,5G,Mondo Smart City Intelligenti -
 ">
">
BANCA DATI: FAGO M13 - Collegato a Morgellons - I computer superveloci del futuro funzioneranno grazie a un virus biologico |
